|
Karamelo
714599e9
Parallel Material Point Method Simulator
|


|
|
Karamelo
714599e9
Parallel Material Point Method Simulator
|
|
This is a very short guide on how to get started with Karamelo.
In order to use Karamelo, you need to download, extract and compile Karamelo's source code. Compiling is no that complicated thanks to the use of CMake (cmake.org). It will automatically check for most dependencies, but unfortunately not all of them.
Karamelo has a few dependencies that need to be satisfied for it to successfully compile and run. They are:
The authors, as many researchers that do simulation work use Linux, and Ubuntu in particular. In order to make it simple for people like us to install and run Karamelo on Ubuntu, here are the steps to follow to download, build, and run the last version of the code. The following steps were tested using the versions 18.04 LTS and 16.04 LTS.
Add the path to Eigen, gzstream and matplotlib to your paths: Add to your .bash_profile:
Remark: change /home/toto/ to the path to the root directory where you have installed gzstream and matplotlib-cpp (they can be different from each other).
Download and install Karamelo:
Here is a simple example of two disks bouncing onto each other to get you started with Karamelo.
This example involves two elastic disks launched at a speed 0.1 mm/s towards each other:
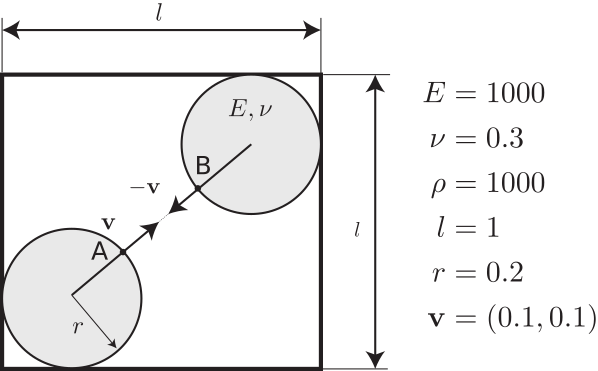
In order to simulate this problem, we need to create an input file that we called two-disks.mpm which reads:
In order to run this example, on 4 CPUs, for instance, simply execute the following command:
Of course, you can change the number of CPU by adjusting the variable NUM_CPU.
This example generates three types of results:
 1.8.13
1.8.13